




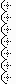



Tuesday, July 22, 2008
 Picture 3: Shows a general overview of how the virus can infect the cell and causes cell lysis. When the cells lysed, it forms a clearing in the plaquing medium. Each clearing can usually be visualized with the naked eyes or using some other techniques such as staining, microscopy, hemadsorption or immunofluorescence. Stain agarose is shown in picture 1 (above)
Picture 3: Shows a general overview of how the virus can infect the cell and causes cell lysis. When the cells lysed, it forms a clearing in the plaquing medium. Each clearing can usually be visualized with the naked eyes or using some other techniques such as staining, microscopy, hemadsorption or immunofluorescence. Stain agarose is shown in picture 1 (above)http://www.flutrackers.com/forum/attachment.php?attachmentid=326&d=1148840493
 Picture 2: Schematic diagram of plaque assay.
Picture 2: Schematic diagram of plaque assay.Retrieved from http://homepages.strath.ac.uk/~dfs99109/BB211/phageinfect.jpg
{kind=link}

Picture 1: Shows a Plaque assay on a 6 well plate. Agarose (or also known as plaquing medium) has been stained with neutral red for easy visualization of plaques. Staining is optional and our lab usually don’t stain it, we just check the number of plaques by holding the plate against the light to check for clearings/plaques.
Retrieved from: http://pathmicro.med.sc.edu/mhunt/plaque.jpg
{kind=link}
To Han Yang
Thanks for reading my entry =) very sorry for the late reply also, didn’t check that there were comments posted for my entry.
1) The type of mammalian cells I’m using is fibroblast but I’m not sure if I can tell you the strain. Sorry about that.
2) The cells that contain the gene I want from step 1 are actually from a company called invitrogen. The purpose of culturing these cells is only to get the gene which was supplied to us as cell cultures, because only these cells is known to contain such genes. The medium we use is according to what was stated in the protocol supplied by the manufacturer. Which was to use normal DMEM with 10% FBS supplemented. I believed the DMEM is a complete medium which already contains essential amino acids, vitamins and salts. Essential amino acids (i.e. those synthesized by the body) are required by most of the culture cells together with cysteine and tyrosine. However, this requirement will vary from one type of cell to another. Probably the amino acids in the DMEM is already sufficient. (I’m not sure but I will confirm with my mentor.)
As for antibiotic, the common and original use of antibiotics is to reduce the frequency of contamination. However, since we are already handling most of our medium in the LAF (laminar air flow hood) and BSC (Biosafety cabinet), there is no need to add antibiotics. We chose not to add antibiotic because the presence of antibiotic in the medium may “hide/mask” the presence of low level contaminations such as bacteria. If there is contamination, we would want to detect it and not use that batch of cultured cells.
3) I do not know why. LOL. =X And my mentor is not around to answer me also. (I will confirm with him when he’s back). Hmmz, but I do have my own answer to this questions. I think the virus titer drops over time because it is being stored in the cold room and used quite often. Although we wrap the tubes with the dull side of the aluminium foil to prevent exposure of the viruses to light (because they are sensitive to light), the opening of the cap still exposes it to light. As for the temperature, I really don’t know. =)
4) Well, he did test the virus efficiency earlier on when he first got the batch of virus produced, but the virus titer can drop over time and we may need to measure it again after about 3-4 weeks. For more detailed understanding of how the lab works read below =)
Batch 1 we call it P1, batch 2 we call it P2, and batch 3 we call it P3 etc. The very first batch of virus we get by following the outline given in the blog is called batch 1 or P1. This batch of virus titer is quite low and if we want to increase/ amplify the titer, we use P1 virus to infect a newly cultured 6 well plate of cells and harvest the virus. This is known as P2 viruses. P2 is supposedly quite concentrated and we do not use this P2 directly. Instead, we do amplify another batch from P2, called P3. This P3 virus is the virus that will be kept in the cold room and used frequently.
We have to produce new P3 stock once P2 virus titer is low. P3 can be used for quite a long time as it has about 20 plus ml, and usually we only need a few hundred µl. Whenever we get a new batch of virus, be it P1, P2 or P3 we will do a plaque assay to get an estimate of the virus titer and then these virus are kept back in the cold room. Without plaque assay we can not know the virus titer.(it takes about 7 days for the results of the plaque assay to be out). Also, the virus titer can drop over time, but sometimes we may have already used the viruses for some of our experiments and if we realized that we did not get the expected results, we may troubleshoot and do plaque assays to check if the virus titer has dropped. However, plaque assay requires quite a lot of cells and since our whole lab share only one flask of cells, maintained in a spinner flask, we can only do plaque assays when we have cells. These cells will only be split on Monday, Wednesday and Friday.
5) Well, since my mentor has dealt with this type of cells before, he already knows roughly the doubling time of the cells. Moreover, the manufacturers who provided the cells also provided the protocol that we should incubate roughly half to one hour to get 50% confluency. The confluency of the cells can be checked under the inverted microscope. 50% confluency means that the cells occupies about 50% of each well of the 6 well plate surface. If it is not 50% confluence yet, we can always return it back into the incubator for the cells to continue to grow. Usually, it should not take more than 1 hour to get 50% confluency for this particular type of cell we are using.
6) Well, the goggle is actually not necessary for the handling of the viruses. We handle the virus in the BSC and should not come into contact with our eyes. The goggle is actually given by the safety department for everyone working in the lab and is recommended that we wear it when we handle with large bottle of chemicals/ liquids, probably TAE buffer when preparing the agarose gel, because of the potential spillage. So I wear my goggles when I am preparing the agarose gel for running my sample. The goggles is required also when we are doing bench work that is handling with a lot of eppendorf tubes, the water droplets on the inner side of the cap may accidentally come into contact with the eyes as we pop open the cap. Also, since I do not wear my specs in the lab, goggles is required.
Jean Leong
TG02
To Ms Chew and Peers
Thanks for reading my entry =)
Plaques are visible structures (usually a clearing as seen in the picture 1) formed in a cell culture contained within a nutrient medium. (e.g the plaquing medium). These clearings has no cells in it because the virus infect the cell and eventually lysing it. (lytic cycle shown in picture 3). The cells that were initially seeded in the well and allow the virus to infect it (from step 1 to 4), then overlay with plaquing medium for the cells to attach to the agarose and grow. (if you use bacteria then a bacteria lawn will be formed;but a different method is used, on agar plate instead of 6 well plate. Done Before in Basic Microbiology. However we do not use bacteria in this case). Just like how each colony is derived from a single E.coli cell in an agar plate, each clearing here is derived from one virus attaching to one cell, and when the virus infects the cell and multiple it continues to lyse the other cells around and forms a clearing around that area. As that clearing, also known as plaque, arise from one virus, we are able to determine the number of viruses (which is also known as titer which is also = no. of plaque forming units/ ml) by counting the number of plaques. =)
1) Harvest the cells that the virus can infect on the 6 well plates and incubate til cells are 50% confluence.
2) Perform serial dilution of the virus such as 10-2, 10-3, 10-4, 10-5, 10-6, 10-7 and 10-8. in eppendorf tubes.
3) Remove medium from the cultured cells and add the diluted virus to the 6 wells and incubate to allow viruses to infect cells.
4) Remove the fluid (containing virus) in each of the well, as the viruses has already attached to the cells.
5) Add plaquing medium, which is a type of agarose gel containing medium to each of the well after the fluid is removed, and allow agarose gel to harden before incubating the plate of cells.
6) The function of the agarose gel is to immobilize the viruses so that it forms a clearing as it infects the cells.
7) These clearings (plaques) can be counted as plaque forming unit/ml, which is the titer of the virus.
Jean Leong
TG 02
=)
10:48 AM