







Hi all,
for the past two weeks, i was attached to the histology lab. It was really a eye-opening experience i saw a lot of interesting things that i had never seen before, like the pathologist sectioning different organs and also observed post-mortem by the professor there. It was rather scary esp for the post mortem done on pre-mature babies.
Yup, but for this entry, i am going to share about frozen section.
What is it?
For frozen section, it is only done for emergency cases. It is done when the patient in the operating theatre and the doctor needs to know if that particular part of the tissue/organ is cancerous or if they have any benign tumors. The doctors would need to know if the margins pf tumors are clear. So what usually happens is they will take out a small section of the part of the tissue and sent to the lab for diagnosis. And as I mentioned they are still in the middle of the operation, they will need the results fast. Therefore, the whole process from the moment the pathologist starts to section to reading the results under the microscope.
When the tissue/section is cut out by the doctors in OT and ready for collection, the staff in the lab will go to the OT to collect the specimen. The section is sent fresh, which means no formalin is used to fix it etc. Tissues are collected in an ice box.
Specimen received must not be sent in formalin, but sent fresh. It will also not be performed for tissue from patients suspected of tuberculosis and other high risk infection like HIV, HBV and HCV etc.
All the patients name, accession number, specimen type, diagnoses are to be recorded down in a handbook for referring purposes. The pathologist will be the one sectioning and there will be a medical technologist to assist them in recording the morphology and dimensions of the specimen sent down. Specimens with history of infection will be handled in the BSC.
All types of tissues can be sent down as long as they are from the human body, usually the more common ones sent down are sections of breast tissue, uterus, gynaecology, cervix and etc.
Procedures
Preparation
-A small amount of O.C.T (optimum cutting temperature) compound is added onto a semi-cold specimen disc. (It is to hold the tissue on the disc and acts as an embedding medium)
- The tissue will be placed on the O.C.T compound
- The tissue will be covered with more O.C.T compound until the entire section is covered.
- It is then quickly placed in liquid nitrogen to freeze the specimen rapidly
- The section disc is inserted into cryocut and the heat extractor cylinder is used to flatten the surface for easier sectioning.
Sectioning
- The frozen section is performed using the Leica Cryocut 1800. The interior of this machine is around -20 to -30 degrees Celsius
- The tissue sections are cut at 5micrometers
- The sectioned specimen were then picked up by using a warm glass slide, the specimen will be attached to the glass slide by itself when it touch the surface of the slide.
- It is then to be stained with Rapid H & E. (used usually as when viewed under the microscope, this stain will give enough contrast and allow better diagnosis)
Staining
- The section fixed in
- 10% formal alcohol for 20s
- Rinse in water
- Haematoxylin for a min
- Rinse in ammonia water
- Wash
- Eosin for 15s
- Dehydrate in 2change of alcohol and 2 change of xylene.
- Mount in DPX
Lastly, the pathologist will view the slides under the microscope and interpret the results. ( to see if there are tumors seen etc.)
That is all =) Hope u enjoy reading
Zhenling
0606970B
TG02
HI, everyone, it’s my turn to blog again. As I am in a research chemistry lab and am not rotated anywhere, thus, for the past 9 weeks, I have been carrying out the same procedures mentioned in my first post (week 4). Thus, for this week’s post, I will be sharing on a procedure that I have observed in my lab.
Technique: Column chromatography
Principle: For separation of a mixture of compounds and isolation of particular component from a mixture
Solvents commonly used (in increasing polarity): Hexane (CH3(CH2)4CH3), dichloro-methane (CH2Cl2), ethyl acetate (CH3COOCH2CH3), acetone (CH3COCH3), methanol (CH3OH).
 Column Chromatography: Image taken from http://orgchem.colorado.edu/hndbksupport/colchrom/colchromproc.html
Column Chromatography: Image taken from http://orgchem.colorado.edu/hndbksupport/colchrom/colchromproc.html In column chromatography, the vertical column is first packed with silica gel (SiO2) that is mixed with hexane. The gel is loaded above a plug of wool. The wool is for preventing solid materials from contaminating the products. The silica gel in the column is known as the stationary phase (also known as the sorbent). After packing the column with the sorbent and loading the sample, another plug of wool is placed above the sample and the sorbent. This is so that later when pouring the solvent in, there is minimum disturbance to the stationary phase and the sample as disturbance to the surface will result in poor separation.
The basic principle is quite similar to the SPE that I have mentioned in my first post. Except that in the SPE I have mentioned in my first post is use C18 (reversed phase) while the column chromatography I have observed use silica gel (normal phase) thus is based on increasing polarity. This means the least polar component of the mixture will be eluted first and the most polar one will be eluted last.
For column chromatography, a very important factor is the choice of solvent for elution for better separation of components. Solvent to use is determined by Thin Layer Chromatography (TLC).
 Thin Layer Chromatography: Image taken from http://www.chemistry.adelaide.edu.au/external/soc-rel/content/tlc.htm
Thin Layer Chromatography: Image taken from http://www.chemistry.adelaide.edu.au/external/soc-rel/content/tlc.htm Using a capillary tube, a drop of the loading solvent containing the sample is applied onto the plate. Then, the plate is placed into the solvent submerging only the bottom of the TLC plate (the location where the sample is spotted.) The solvent will then slowly migrate up the plate. It is a matter of trial and error of different kinds of solvents with different ratio to find the ideal solvent to be used for column chromatography. The desired outcome is for the desired spot (the compounds we want) to be as near to the baseline as possible. Baseline is the location where the drop of sample is placed at.
Why is this the desired outcome?
Why in TLC the spot nearest the baseline is the most polar?
So in the case of the desired spot being too far away from the baseline, a less polar solvent is used to bring the spot closer to the baseline.
How to get a less polar solvent?
If the compounds are colourless, iodine vapour is then used to visualise the spots. This is done by dipping the plate into a bottle of iodine (powder form). Why use iodine in solid form? If recall from secondary school chemistry, pure iodine at room temperature is in solid state, iodine can sublimes at room temperature.
After finding out what solvent to use as the eluent, next we had to check whether the loading solvent is more or less polar than the eluent. If the eluent is more polar than the loading solvent, the eluent can be used directly in the column chromatography to remove the loading solvent. If the eluent is less polar than the loading solvent, a less polar solvent such as hexane is loaded first before loading the eluent. This is because if the loading solvent is more polar than the eluent, it will function as the eluent and push the sample forward, thus the sample will travel over a shorter distance, which will result in less separation.
In column chromatography, since the compounds are colourless, we will not know to collect till which mark, thus the eluent are collected in a series of small tubes. Thus TLC is carried out on each of the tubes with a drop of the desired pure standard beside. A spot that is at the same height as the pure standard with no other spots elsewhere indicates the particular tube where the sample is taken from is pure with only the desired compound in it. Next, all the samples which are pure with only the desired compound in it are combined.
Ta-ta…..pure desired compound is obtained from the mixture.
Feel free to ask questions.
Hello folks! My turn 4 posting again:D
I'll keep it short and sweet k :D
Past 4 weeks i've been in cytogenetics and i learnt alot on chromosomes and their abnormalities.
In this posting i'll talk bout Wolf Hirschhorn Syndrome.
Wolf Hirschhorn Syndrome is causes by a genetic error in the 4th chromosome. The short arm of chromosome 4 has a partial deletion. This could be inherited from the parent chromosome where there is a translocation. Wolf hirschhorn is also known as 4p- syndrome because the deletion is in the p arm of the chromosome.
 Retrieved on 17 August 2008 from, http://www.slh.wisc.edu/cytogenetics/cases/gifs/com_karyotypes/CoMMar97karyo.gif
Retrieved on 17 August 2008 from, http://www.slh.wisc.edu/cytogenetics/cases/gifs/com_karyotypes/CoMMar97karyo.gif
{kind=link}
Wolf hirschhorn syndrome affects fetal growth and developement, hence causing malformations in most body parts.
Facial features are described ad the "Greek warrior helmet" features. Their forehead is usually prominent with wide eyes and broad beaked nose. They are short in stature and have malformations of hands, feet and spine. Likely to have heart defects and malformations or underdevelopment of organs(urinary and genitals).Profound mental retardation, small head and seizures(50% of the individuals are affected) are the brain and muscular features of wolf hirschhorn symdrome.
Retrieved on 17 august 2008, from, http://medgen.genetics.utah.edu/photographs/diseases/high/17_mod.jpg
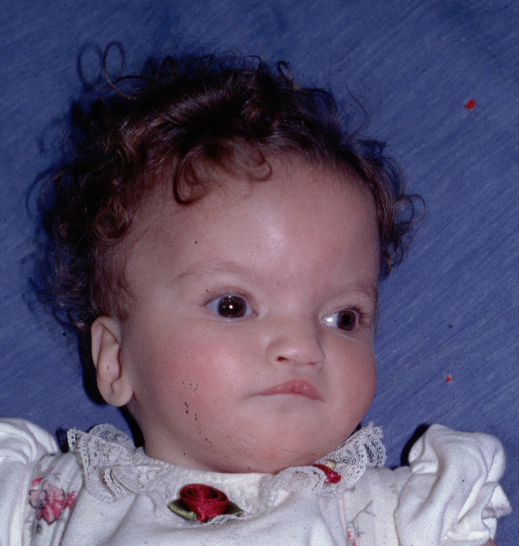{kind=link}
Wolf Hirschhorn syndrome could be detected by fluorescence in-situ hybridisation(FISH). It detects the deleted portion of the 4p chromosome by molecular probes which fluoresces. Addditional tests could include x-rays to look for bone and internal malformations, renal ultrasonography to examine the kidneys and magnetic resonance for the imaging of brain.
Thats all for this posting. i guess i've explained very briefly, so don't hesitate to ask questions.
Thanks:D
Neela
TG02
hmm, okayy, i'm next! haha. anyway, i know i posted late again, thats cause i was in jakarta. sorry to all.
Anyway, this is about my experience that things that i have learnt in processing for the past 2 weeks and hopefully more to come for the next 2 and a 1/2 weeks. haha.
Processing - the 'general office' in the lab as every sample received from the clinics or the wards are processed by the lab clerks first before being dispatched out to the respective sections such as routine / haematology.
I had to be the 'runner' when the canister tubing system was being upgraded. haha. great exercise for the legs. haha. But luckily, our supervisor decided to make the porters (not sure bout the spelling) come in the lab instead and i got my own 'coffee table' where i got to sort out the awaiting and non awaiting samples, the micrology, histology and routine samples. It can get really busy and messy at times.
Next, i had to be a paster, by sticking the ID labels on the order forms and the blood/urine /stool samples. It is crucial to check that the name on the specimen tallies with the order form as errors do happen. At times, empty or insufficient specimen bottles and unlabelled specimens are received and its is quite troublesome. However, as a student, i won't be able to be the one to call the nurse/wards to correct the issue. haha. (Its a good thing though.)
Then, for the most days of the past 2 weeks, fatin, elyn and i were up to our necks with the preparation of samples for the Health Awareness Week that is currently ongoing. On one of the days, we had to work till 7.30 p.m as we had to send the forms and the tubes to the respective wards. It was quite fun though.
Anyway, my job in processing is quite self explanatory but nevertheless, feel free to ask me anything. =)
Debbie.
Tg02
Hi everyone, it is my turn to share again =) Remember the first posting on the construction of the virus? Well, that is just a brief outline of what I have been doing for the past 6 weeks and will continue to be doing for the next few weeks.
*(Later I will share with you why it takes such a long time) ==> remember this one of the big question for this post, if you can answer this, then you did a *good job in understanding this entry =) heex. Those who cannot..(no such thing k) lols.jk. can ask me =)
Basically, because I am attached to a research lab, I planned to share more of some of the techniques the labs are using nowadays for molecular biology research work and some cell culturing techniques in the later posts.
I will break down the procedures into a few separate postings so that more detailed learning of each of the techniques can be shared.
For the procedure in the construction of virus it involves two MAJOR components.
1. Cloning
2. Packaging into the virus (I haven reach here! so this will be shared next next time k, because I foresee first step will take very long==> why? Find out by continue reading..)
1. Cloning
Cloning in this context refers to isolating a defined DNA sequence and obtaining multiple copies of it. Cloning is use to amplify (by using living cells such as bacteria E.coli) the specific DNA sequence containing the (gene of interest)*secret.
E.coli is often used because of the easy manipulation and it can produce high copies of plasmid.
(the gene of interest)*
E.g. Gene A ==> has to be cloned==> packaged into the virus==> get virus A==> introduce gene into mammalian cells
E.g. Gene B==> has to be cloned==> packaged into the virus==> get virus A==> introduce gene into mammalian cells
Each (gene of interest) has to be clone separately because each gene size is very big: up to 2 to 2.5kb.The longer the sequence of the gene the easier/ higher possibility of getting mutation in the gene when we are doing cloning. This is because during the procedure of cloning, it involves a lot of manipulation of the gene, especially PCR which can introduce single bp mutations. Even if very high fidelity (means accuracy of the copy to the source) PCR mix is used, the PCR process is not 100% perfect.
Bacterial cells containing the gene of interest is provided by the manufacturer in a very interesting form. It is in a small transparent glass bottle (cylinder shape) which has ¼ filled agar inside, and one very big white colony is seen sitting on the agar. (The bottle is only about 4cm in height and 1.5cm in diameter). The manufacturer provided it in the bacteria form on agar for easy usage, because we can just pick and use the bacteria.
Cloning can be further broken down into 4 main parts- this is called the “Cloning Strategy”
1(a) Isolation of DNA insert
1(b) Ligation
1(c) Transformation
1(d) Screening (Blue-white) / Selection strategy (antibiotics)
According to the procedures shared in post 1, this is a summary map of the procedures. (Construction of virus: cloning and packaging into the virus).
You CAN click on the picture

Picture taken from http://www.invitrogen.com
I am currently still in the green box, which means I have gone through these steps:
1(a) Isolation of DNA insert ==> Original (gene of interest) purchased from manufacturer, that comes in the form of plasmid contained in bacteria cells.
1(b) Ligation ==> of the (gene of interest) into Vector Backbone containing 7nTR (Facilitate later steps)==> to get a recombinant plasmid.
1(c) Transformation==> of recombinant plasmid into first type of E.coli cells (lets call it 5a)
1(d) Selection strategy ==> of the transformed cells (5a) (using amphiciliin antibiotic)==> then pick a one colony randomly *clue and culture it in broth *(15hours) ==> extract the recombinant plasmidè restriction digestion to cut out gene of interest and run on gel to confirm presence of gene of interest ==> and send for sequencing (using the fluorescence labeled ddNTP) to check for any mutation of the (gene of interest) IN THE recombinant plasmid ==> if there is mutation of the bp (seen in sequencing results) ==> go back to the plate and *re pick the colony, re grow them and sequence again. (thats partly why my lab people always say "pray hard!"==> cos its like kinda 'chance' thing even when optimal conditions are provided.)
Cloning process can take up to 1month if every goes well, but more than 1 month if we keep re picking the colonies. Usually, we will number the colonies on the agar plate, so that we know if that colony has already been picked before.
Sequencing process is done by sending it to an external company which does sequencing, E.g. The company 1st base. We have to provide the samples (picked from colony) and primers specific to the gene we want to sequence. Usually sequencing results using the fluorescent ddNTP method starts to become less sensitive when it reaches the later part (~700bp), therefore one primer for every 700bp we want to sequence should be provided). Sequencing *results usually takes about 3 to 4 days to arrive and usually a soft and a hard copy will be provided. The soft copy shows both the sequence in e.g. “actgactg form” and the electropherogram, while the hard copy shows us the electropherogram results with different coloured peaks but the different bases (a, t, g, c) only.
In the green box, it shows a recombinant plasmid which I am supposed to get to proceed to transformation of second type of E.coli cells (lets call it 10a) ==> shown in the picture named as “competent E.coli cells”.
Steps for :
Isolation of DNA insert
Therefore once I received the bacteria cells (gene of interest inside) ==> plate them on LB +amp plate to get single colony==> pick one colony==> grow in broth (15hours) ==>extract the DNA è PCR to amplify the gene of interest and introduce restriction sites è restriction digestion ==>
Ligation, transformation and selection
Ligation==> transformation ==> selection==> pick colonies ==> grow them in broth (15hours) ==> extract the DNA ==> restriction digestion to cut out the insert (confirm insert is inside recombinant plasmid) ==> sequencing (using the fluorescence labeled ddNTP)
Most of the techniques are just recapped from the molecular genetics and molecular Biology and culturing of bacteria from basic microbiology.
One thing which I found very interesting is this technique: Cloning of PCR product, I ask the other students and a lot people never heard of this before in school! so it’s a good thing to know =)
PCR refers to polymerase chain reaction, it is a process which allows the amplification of any region or even very complex genomes in just a few hours. (for details refer to link)
http://bcs.whfreeman.com/lodish6e/default.asp?s=&n=&i=&v=&o=&ns=0&uid=0&rau=0
go to this website, click on animation and then PCR
Cloning of PCR product
PCR product can be directly used for coloning? How?
A restriction enzyme recognition site can be added to the 5’ end of the oligonucleotide primers used for the PCR reaction. These sequences will also be into corporated into the amplified PCR product as the PCR cycles continue, it can later be digested to produce the specific sticky or blunt ends for ligation. The primer will bind to the complementary part of the DNA template, but the restriction enzyme recognition site does not match the template. However, since the direction of synthesis occurs in the 5’ to 3’ direction and specificity depends mainly on the 3’ end of the primer, the DNA still can be amplified efficiently and the product will contain the restriction sites at its end. Extra bases are also eventually and normally added to the 5’ end of the restriction site to ensure that the restriction enzymes functions efficiently.
Taken from PCR by (1997) second edition from C.R. Newton & A.Graham
Hope you learnt new things! =) Take care!
Jean Leong
TG02
0607991G
Reference
1. C.R. Newton & A.Graham.(1997) PCR (2nd edition). BIOS Scientific Publishers Ltd, USA.
{kind=link}